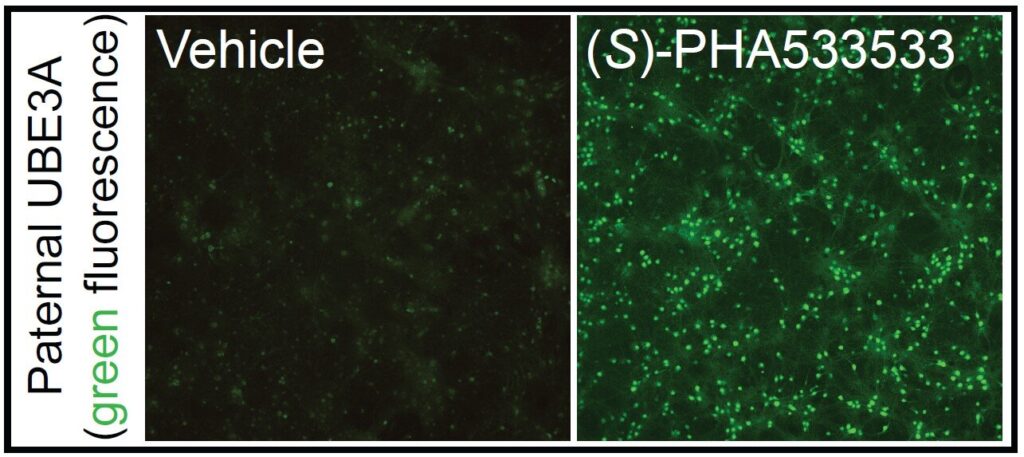
Les neurones porteurs d’un rapporteur fluorescent pour l’activité du gène paternel UBE3A ne présentent qu’une faible fluorescence de fond lorsqu’ils sont traités avec un véhicule témoin, mais présentent une fluorescence brillante lorsqu’ils sont traités avec (S)-PHA533533, ce qui indique que la petite molécule a puissamment activé l’allèle paternel dormant d’UBE3A. Crédit : Vihma et al 2024
Le syndrome d’Angelman est une maladie génétique rare causée par des mutations du gène UBE3A hérité de la mère et caractérisée par un mauvais contrôle musculaire, une élocution limitée, une épilepsie et des déficiences intellectuelles. Bien qu’il n’existe pas de remède contre cette maladie, de nouvelles recherches menées à la faculté de médecine de l’UNC ouvrent la voie à un tel traitement.
Ben Philpot, Ph.D., professeur distingué Kenan de biologie cellulaire et de physiologie à la faculté de médecine de l’UNC et directeur associé du centre de neurosciences de l’UNC, et son laboratoire ont identifié une petite molécule qui pourrait être sûre, délivrée de manière non invasive et capable d’« activer » la copie dormante du gène UBE3A hérité du père dans tout le cerveau, ce qui conduirait à un bon fonctionnement des protéines et des cellules, équivalant à une sorte de thérapie génique pour les personnes atteintes du syndrome d’Angelman.
« Le composé que nous avons identifié s’est révélé très bien absorbé par les cerveaux en développement des modèles animaux », a déclaré Philpot, un éminent spécialiste du syndrome d’Angelman. « Il nous reste encore beaucoup de travail à faire avant de pouvoir lancer un essai clinique, mais cette petite molécule constitue un excellent point de départ pour développer un traitement sûr et efficace contre le syndrome d’Angelman. »
Mais ces résultats, qui ont été publiés dans Nature Communicationsmarque une étape majeure dans le domaine, selon Mark Zylka, professeur distingué WR Kenan Jr. de biologie cellulaire et de physiologie à la faculté de médecine de l’UNC et directeur du centre de neurosciences de l’UNC. Aucun autre composé à petite molécule n’a encore montré autant de promesses pour Angelman, a-t-il ajouté.
Contrairement à d’autres maladies monogéniques telles que la fibrose kystique et la drépanocytose, le syndrome d’Angelman présente un profil génétique unique. Les chercheurs ont découvert que les enfants atteints de ces maladies n’ont pas la copie du gène UBE3A héritée de la mère, tandis que la copie du gène UBE3A héritée du père reste latente dans les neurones, comme c’est le cas chez les individus neurotypiques.
En règle générale, l’UBE3A aide à réguler les niveaux de protéines importantes ; l’absence d’une copie fonctionnelle entraîne de graves perturbations dans le développement du cerveau.
Pour des raisons qui ne sont pas entièrement claires, la copie paternelle du gène UBE3A est normalement « désactivée » dans les neurones de tout le cerveau. Ainsi, lorsque la copie maternelle du gène UBE3A est mutée, cela entraîne une perte de la protéine UBE3A dans le cerveau. Philpot et d’autres chercheurs ont émis l’hypothèse que l’activation de la copie paternelle du gène UBE3A pourrait aider à traiter cette maladie.
Hanna Vihma, Ph.D., chercheuse postdoctorale au laboratoire Philpot et première auteure de l’étude, et ses collègues ont examiné plus de 2 800 petites molécules d’une bibliothèque chimiogénétique de Pfizer pour déterminer si l’on pouvait activer puissamment l’UBE3A paternel dans des modèles de souris atteints du syndrome d’Angelman.
Les chercheurs ont modifié génétiquement les cellules neuronales de souris avec une protéine fluorescente qui s’allume lorsque le gène UBE3A paternel est activé. Après avoir traité les neurones avec plus de 2 800 petites molécules pendant 72 heures, les chercheurs ont comparé leurs milliers de cellules traitées à celles traitées avec du topotécan, une petite molécule connue pour activer le gène UBE3A paternel, mais qui n’a pas de valeur thérapeutique dans les modèles animaux de la maladie.
Le (S)-PHA533533, un composé précédemment développé comme agent antitumoral, a provoqué l’expression par les neurones d’une lueur fluorescente rivalisant avec celle induite par le topotécan, ce qui signifie que son effet était suffisamment puissant pour activer avec succès l’UBE3A paternel.
Les chercheurs ont pu confirmer les mêmes résultats en utilisant des cellules souches pluripotentes induites dérivées d’humains atteints du syndrome d’Angelman, indiquant que ce composé a un potentiel clinique.
De plus, les chercheurs ont observé que le (S)-PHA533533 présente une excellente biodisponibilité dans le cerveau en développement, ce qui signifie qu’il se déplace facilement vers sa cible et y reste. Ceci est remarquable dans la mesure où les thérapies génétiques précédentes pour le syndrome d’Angelman avaient une biodisponibilité plus limitée.
« Nous avons déjà montré que le topotécan, un inhibiteur de la topoisomérase, avait une très faible biodisponibilité dans des modèles murins », a déclaré Vihma. « Nous avons pu montrer que le (S)-PHA533533 avait une meilleure absorption et que la même petite molécule pouvait être traduite dans des cellules neuronales d’origine humaine, ce qui est une découverte majeure. Cela signifie que ce composé, ou un composé similaire, a un véritable potentiel comme traitement pour les enfants. »
Bien que le (S)-PHA533533 soit prometteur, les chercheurs s’efforcent toujours d’identifier la cible précise à l’intérieur des cellules qui provoque les effets souhaités du médicament. Philpot et ses collègues doivent également mener d’autres études pour affiner la chimie médicinale du médicament afin de garantir que le composé – ou une autre version de celui-ci – soit sûr et efficace pour une utilisation future en milieu clinique.
« Il est peu probable que ce soit exactement le composé que nous utiliserions pour la clinique », a déclaré Philpot. En collaboration avec les chimistes médicinaux du laboratoire du Dr Jeff Aubé, le laboratoire Philpot s’efforce d’identifier des molécules similaires présentant des propriétés médicamenteuses et des profils de sécurité améliorés. « Cependant, cela nous donne un composé avec lequel nous pouvons travailler pour créer un composé encore meilleur qui pourrait être utilisé pour la clinique. »
Plus d’information:
Unsilencer Ube3a pour le traitement potentiel du syndrome d’Angelman, Nature Communications (2024). DOI: 10.1038/s41467-024-49788-8
Fourni par le service de santé de l’Université de Caroline du Nord
Citation: Des chercheurs identifient une molécule comme traitement potentiel du syndrome d’Angelman (2024, 8 juillet) récupéré le 8 juillet 2024 à partir de
Ce document est soumis au droit d’auteur. En dehors de toute utilisation équitable à des fins d’étude ou de recherche privée, aucune partie ne peut être reproduite sans autorisation écrite. Le contenu est fourni à titre d’information uniquement.